Idiopathic inflammatory muscle disease causing symmetric proximal muscle weakness due to T-cell–mediated muscle fiber (endomysial) inflammation.
A treatable cause of acquired muscle weakness in adults and a classic autoimmune myopathy tested on boards. Polymyositis can significantly impair mobility and may be associated with interstitial lung disease or other autoimmune disorders. It also requires differentiation from look-alikes (dermatomyositis, inclusion body myositis, etc.).
Gradual onset (weeks-months) of symmetrical proximal weakness (difficulty climbing stairs, rising from a chair, combing hair). No rash or skin changes (unlike dermatomyositis).
Usually affects adults in their 40s-60s (more common in females). Patients may have muscle aching but weakness predominates; reflexes are generally intact (unless severe atrophy).
May have dysphagia (if esophageal muscles involved) or mild systemic symptoms (fatigue, low-grade fever, weight loss). In some cases, associated interstitial lung disease (especially with anti-Jo-1 antibody) causing dry cough and shortness of breath.
Labs: check CK (typically very high, often >10× normal), aldolase, AST/ALT (muscle enzymes) to confirm myopathic injury.
Rule out mimics: test TSH (exclude hypothyroid myopathy) and review medications (e.g., statins, steroids) that can cause myopathy without inflammation.
Autoantibodies: screen for ANA (positive in ~80% of cases); if positive, check myositis-specific antibodies like anti-Jo-1 (antisynthetase) or anti-SRP.
EMG (electromyography): supports diagnosis by showing myopathic changes (irritable muscles with fibrillations, short-duration motor unit potentials).
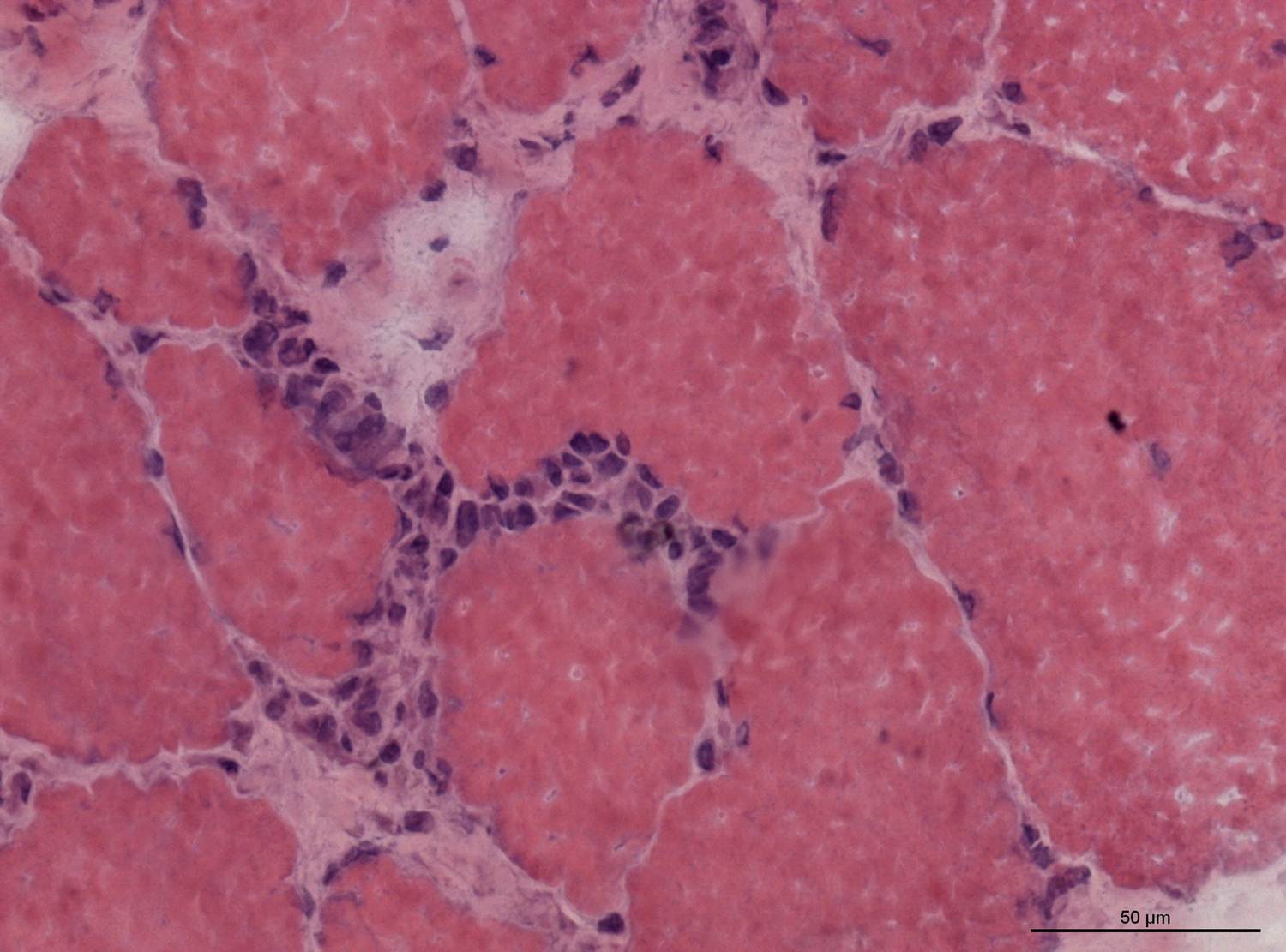
Muscle biopsy: definitive test showing endomysial inflammatory infiltrates (CD8+ T-cells) with muscle fiber necrosis; necessary if diagnosis is uncertain.
in older adults, causes proximal muscle pain/stiffness (not true weakness) with normal CK; associated with giant cell arteritis
High-dose corticosteroids (e.g., prednisone ~1 mg/kg/day) are first-line; prolonged taper as strength improves. Most patients respond to steroids (improved muscle strength over weeks).
Add a steroid-sparing immunosuppressant (methotrexate or azathioprine) if needed for inadequate response or to reduce long-term steroid use. IVIG can be helpful in refractory cases (especially for severe weakness or dysphagia).
Supportive care: physical therapy to regain muscle strength and prevent atrophy; screen and treat extra-muscular involvement (e.g., pulmonary function for ILD). Ensure age-appropriate cancer screening (underlying malignancy is less common than in dermatomyositis but still possible).
Polymyositis is a T-cell (CD8)-mediated attack on muscle fibers from within (endomysial inflammation) — in contrast, dermatomyositis involves CD4+ cells and perifascicular inflammation.
CK is usually massively elevated in polymyositis (often thousands); by contrast, polymyalgia rheumatica has normal CK despite similar aches and stiffness.
Anti-Jo-1 antibody (anti-tRNA synthetase) is classic for polymyositis and indicates the antisynthetase syndrome (myositis + ILD + arthritis + "mechanic's hands").
Severe dysphagia or neck muscle weakness → risk of aspiration pneumonia; monitor swallowing function and consider temporary feeding support if needed.
Polymyositis in an older patient with atypical features → evaluate for an occult malignancy (while malignancy association is stronger in dermatomyositis, screening is advised in polymyositis too).
Symmetric proximal muscle weakness (no rash) → suspect polymyositis.
Check muscle enzymes (↑CK, aldolase) and basic labs; exclude other causes (TSH for thyroid, medication review).
If muscle enzymes are elevated, test for myositis autoantibodies (ANA, anti-Jo-1) and perform EMG.
Confirm diagnosis with muscle biopsy showing endomysial inflammation and fiber necrosis.
Start high-dose steroids; add immunosuppressants if needed. Assess for complications (ILD, dysphagia) and consider malignancy screening.
Middle-aged woman with progressive difficulty climbing stairs and combing hair, very high CK, positive anti-Jo-1, and no skin rash → polymyositis.
Patient with polymyositis who has rough, cracked hands ("mechanic's hands") and new-onset cough/dyspnea → suggests antisynthetase syndrome (polymyositis with interstitial lung disease, typically anti-Jo-1 positive).
Case 1
A 50‑year‑old woman presents with 3 months of increasing difficulty climbing stairs and lifting objects over her head.