Autosomaldominant muscular dystrophies (DM1 & DM2) caused by unstable repeat expansions; defined by progressive muscle weakness (distal in DM1 vs proximal in DM2), myotonia (delayed muscle relaxation), and multi-system involvement (early cataracts, cardiac arrhythmias, etc.).
Most common adult-onset muscular dystrophy (≈1:8000). Often underdiagnosed for years due to subtle early signs. Important as a model for anticipation (worsening in each generation) and a cause of life-threatening cardiac conduction issues (risk of sudden death). Frequently tested due to its classic trinucleotide repeat mechanism and multi-system features.
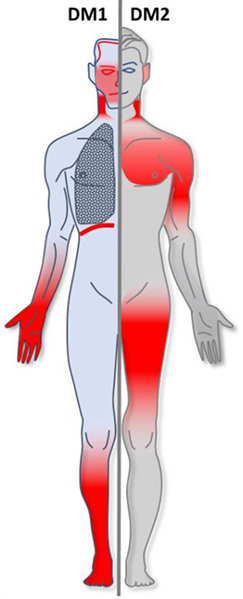
Classic DM1 (Steinert disease): Onset in teens–30s with facial and distal muscle weakness (e.g., foot drop, hand grip difficulty). Look for grip myotonia (trouble releasing a handshake or doorknob) and atrophy of facial muscles (ptosis, hollowed cheeks → "hatchet face"). Systemic features include frontal balding, early cataracts (often <50 years), testicular atrophy in males, and arrhythmias (palpitations, syncope). Severity ranges from mild (just cataracts & myotonia) to severe disability; a congenital form (usually maternal transmission) causes neonatal hypotonia, respiratory failure, and intellectual disability.
DM2 (proximal myotonic myopathy): Onset typically in 30s–50s. Generally milder than DM1 with predominantly proximal muscle weakness (difficulty climbing stairs or combing hair) and muscle pain/stiffness. Myotonia is present but often less pronounced than in DM1. Facial muscles are usually spared or only mildly involved. Systemic findings (if present) may include cataracts and cardiac conduction defects, but DM2 lacks a congenital form and shows minimal anticipation.
Overlapping features: Both DM1 and DM2 are multisystem disorders. Beyond muscle weakness and myotonia, patients can have daytime sleepiness, insulin resistance or diabetes, thyroid dysfunction, and gastrointestinal issues. Cardiac conduction block (AV node dysfunction) is a dangerous complication in both types. Family history is often positive (autosomal dominant). Any young adult with unexplained muscle weakness, early cataracts, and difficulty relaxing muscles should prompt consideration of a myotonic dystrophy.
Clinical clue is myotonia on exam (grip or percussion myotonia) in a patient with pattern of muscle weakness; confirm by EMG which shows characteristic myotonic discharges (waxing and waning potentials).
Genetic testing is the definitive diagnostic test: PCR (and possibly Southern blot) detects the abnormal CTG repeat expansion in the *DMPK* gene for DM1 or CCTG expansion in the *CNBP* gene for DM2. Nearly 100% of affected individuals have an expansion if tested.
Differentiate from other myopathies: CK levels in DM are normal or mildly elevated (hundreds) unlike the very high levels in inflammatory myositis. The presence of myotonia and multisystem features (cataracts, endocrinopathies) distinguishes DM from limb-girdle muscular dystrophies or ALS (which has upper motor neuron signs and no myotonia).
Evaluate cardiac and other systems at diagnosis: obtain ECG (look for AV block or arrhythmias) and echocardiogram, check fasting glucose and TSH, and perform a baseline eye exam. Periodic surveillance is needed (e.g., annual ECG, glucose).
Counsel patients on avoiding triggers that worsen muscle weakness (e.g., certain anesthesia meds, cold exposure for myotonia). Advise genetic counseling for family planning, especially since DM1 can worsen in the next generation (anticipation).
Condition
Distinguishing Feature
Limb-girdle muscular dystrophy
proximal weakness without myotonia or multisystem involvement; often autosomalrecessive
Amyotrophic lateral sclerosis (ALS)
motor neuron disease with muscle wasting but also upper motor neuron signs (hyperreflexia, spasticity); no myotonia or cataracts
Myotonia congenita
isolated myotonia (delayed relaxation) from a chloride channel mutation; presents in childhood with muscle stiffness but no weakness or systemic features
No cure yet; management is supportive and focuses on specific manifestations. Encourage regular exercise to maintain muscle strength and mobility.
Myotonia can be treated if functionally limiting: e.g., mexiletine (a sodium channel blocker) often reduces myotonic stiffness. Other options include anticonvulsants (lamotrigine, etc.).
Monitor and manage systemic issues: e.g., yearly EKG (consider pacemaker if AV block or significant arrhythmia), surgical removal of cataracts when vision is affected, oral hypoglycemics/insulin for diabetes, thyroid hormone for hypothyroidism, and hormone replacement for hypogonadism. Use ankle-foot orthoses or mobility aids for weakness as needed, and ensure respiratory support if ventilatory failure develops.
Mnemonic for DM1: "Myotonic" = My Tonia, My Testicles, My Toupee, My Ticker (myotonia, testicular atrophy, frontal baldness, arrhythmia) – highlights the classic features of Steinert disease.
Repeat expansions display anticipation (worse and earlier in successive generations). DM1 is known for this (e.g., congenital cases typically have an affected mother), whereas DM2 shows little to no anticipation.
DM1 = 1st (distal) muscles affected; DM2 = 2nd (proximal) muscles affected – helps recall the distribution of weakness in each type.
Episodes of syncope or palpitations in DM patients → suggests conduction block or arrhythmia (risk of sudden cardiac death). These findings warrant urgent cardiology evaluation for possible pacemaker/ICD.
Care with general anesthesia: patients with myotonic dystrophy are prone to intraoperative and postoperative complications (arrhythmias, prolonged muscle paralysis). Always alert the anesthesia team to the diagnosis so they can avoid problematic medications (e.g., succinylcholine).
Suspect myotonic dystrophy in a patient with characteristic myotonia (delayed muscle relaxation) plus progressive weakness (distal or proximal) and/or early cataracts or family history of similar illness.
Initial workup: perform an EMG to confirm myotonic discharges; check CK (often normal or mildly elevated) and basic labs to exclude other causes (e.g., TSH for hypothyroid, inflammatory markers for myositis).
Confirm diagnosis with genetic testing: PCR for CTG repeat in *DMPK* (DM1) or CCTG repeat in *CNBP* (DM2). A positive test establishes the diagnosis definitively.
Upon diagnosis, assess disease extent: obtain an ECG (screen for heart block or arrhythmia) and an echocardiogram, check pulmonary function if respiratory symptoms, and screen for metabolic issues (glucose tolerance, thyroid levels). Perform an eye exam for cataracts.
Long-term management: involve an interdisciplinary team. Ensure regular cardiac follow-up (yearly ECG ± Holter), endocrine monitoring (glucose, thyroid), and ophthalmologic exams. Address symptoms with appropriate therapies (pacemaker, medications for myotonia, etc.), and provide genetic counseling.
A 25‑year‑old man cannot release his grip after a handshake (delayed relaxation), has bilateral ptosis and temporal muscle wasting, early balding, and a father who had the same condition → Myotonic dystrophy type 1 (Steinert disease).
A 45‑year‑old reports years of muscle aches and progressive difficulty climbing stairs. Exam shows proximal leg weakness and mild myotonia; workup reveals early cataracts. Initially misdiagnosed as polymyositis, but negative inflammatory markers → Myotonic dystrophy type 2.
An infant is born limp and not breathing well. The baby has severe hypotonia and clubfoot. The mother has muscle weakness and grip myotonia on exam → Congenital myotonic dystrophy (DM1) (extreme repeat expansion from maternal transmission).
Case 1
A 28‑year‑old woman is evaluated for difficulty releasing her grip after using tools and progressive weakness in her hands and feet.
Case 2
A 50‑year‑old man has chronic muscle pain and stiffness. He notes trouble rising from chairs over the past few years.
Areas of muscle involvement in myotonic dystrophy: DM1 (red highlights in face/neck, forearms, hands, lower legs) vs DM2 (red highlights in shoulders, upper arms, thighs).