Genetic disorders characterized by numerous fluid-filled cysts in the kidneys. Autosomaldominant PKD (ADPKD) is common (≈1:500) and usually presents in adulthood (mutations in PKD1 or PKD2); Autosomal recessive PKD (ARPKD) is rare (≈1:20,000) and presents in infancy (mutation in PKHD1).
ADPKD is a significant cause of end-stage renal disease (responsible for ~10% of dialysis patients) and has systemic implications (liver cysts, aneurysms). It often appears in exams highlighting dominant inheritance or kidney cysts with berry aneurysm. ARPKD, while rare, is high-yield for its often fatal neonatal presentation and lifelong complications in survivors (kidney and liver failure).
ADPKD (adult form): Young adult with early-onset hypertension, flank pain, and recurrent hematuria or UTIs. Often has a family history of kidney disease. Kidneys are enlarged (sometimes palpable); extrarenal findings include liver cysts and cerebral aneurysms.
ARPKD (infantile form): Newborn or infant with massively enlarged echogenic kidneys, respiratory distress from pulmonary hypoplasia (Potter sequence), and signs of congenital hepatic fibrosis (portal hypertension, hepatosplenomegaly). If milder, presents in childhood with growth failure and hypertension.
Suspect PKD in patients with family history of kidney cysts or unexplained hypertension with enlarged kidneys.
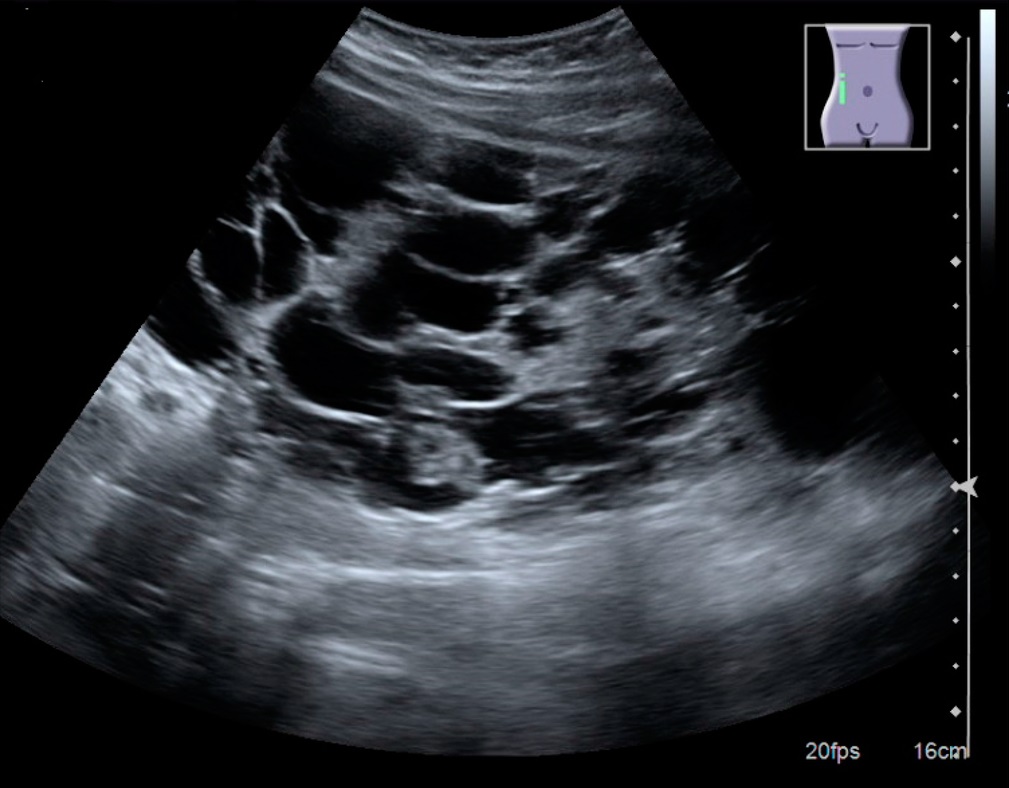
Confirm diagnosis with renal ultrasound (first-line): multiple bilateral cysts on imaging (criteria vary by age). Use CT/MRI if ultrasound is equivocal or to assess total kidney volume.
Consider genetic testing for PKD1/PKD2 (ADPKD) or PKHD1 (ARPKD) especially if ultrasound is inconclusive or for early screening in at-risk individuals.
Evaluate kidney function (baseline creatinine, eGFR) and monitor over time. Control blood pressure (helps slow cyst progression) and avoid nephrotoxic drugs.
Screen for complications: e.g., intracranial aneurysm (MRA) if family history of aneurysm or high-risk factors; monitor for UTIs (which may indicate infected cyst) and treat promptly.
Condition
Distinguishing Feature
Simple renal cysts
few in number, common in older adults; incidental findings without kidney enlargement
Acquired cystic kidney disease
multiple cysts in patients with long-standing dialysis (not inherited; risk of renal tumors)
Tuberous sclerosis complex
has kidney cysts + benign tumors (angiomyolipomas), with seizures and skin findings (ash-leaf spots)
Aggressive blood pressure control (target <130/80 with ACE inhibitor) to slow cyst growth; encourage high fluid intake (to suppress ADH).
For rapidly progressing ADPKD, consider tolvaptan (vasopressin V2 receptor antagonist) to slow cyst development (monitor liver enzymes while on therapy).
Manage complications: prompt antibiotics for cyst infections (fluoroquinolones penetrate cysts), pain management (avoid NSAIDs; consider cyst aspiration or surgical decompression if needed), and hydration to prevent kidney stones.
Plan for renal replacement in advanced disease: dialysis when kidneys fail and ultimately kidney transplant (transplant is definitive; combined liver-kidney transplant if severe hepatic fibrosis).
Mnemonic: 'polycystic kidney' has 16 letters → PKD1 on chromosome 16; PKD2 is on chromosome 4.
ADPKD's extrarenal features: cysts in liver (common), pancreas, spleen, and risk of berry aneurysms in the brain.
Sudden, severe headache in an ADPKD patient → evaluate for ruptured berry aneurysm (subarachnoid hemorrhage emergency).
In ARPKD, neonatal respiratory distress from massive kidneys → may require immediate ventilation support and even neonatal nephrectomy to relieve lung compression.
Family history of PKD or multiple kidney cysts on imaging → suspect PKD.
Perform kidney ultrasound: if age-based cyst criteria are met, confirm the diagnosis.
If ultrasound is negative but suspicion remains (e.g., young patient with affected parent), do genetic testing or follow up later (cysts develop with age in ADPKD).
Differentiate subtype: ADPKD (autosomal dominant) usually in adults with affected parent; ARPKD (autosomal recessive) in infants/children with unaffected parents.
Once diagnosed, manage with blood pressure control and monitoring. Screen for complications (aneurysm, infection) and prepare for dialysis/transplant as disease progresses.
30‑year‑old with hypertension, recurrent hematuria, bilateral flank masses, and a family history of kidney failure → Autosomal dominant PKD (ADPKD).
Newborn with large, bilateral flank masses, pulmonary insufficiency at birth (oligohydramnios), and later portal hypertension → Autosomal recessive PKD (ARPKD).
Case 1
A 35‑year‑old man with a history of hypertension and intermittent hematuria is evaluated for flank pain.
Ultrasound of a polycystic kidney with many large cysts.